About fragments and reads
In NGS bioinformatics everything starts with reads. However reads comming from different sequencing platforms have different definitions. But before I discuss these read definitions, I’ll start biological.
When DNA or RNA is getting preped in the lab, these are sheared (cut into pieces). These pieces of DNA (or cDNA in case of RNAseq) are called fragments. In some protocols, this shearing is random, so fragments start randomly. In other protocols, these fragments start with a certain sequence (like with enzyme recognition sites, in case of GBS or RAD protocols, or with primer sequence, in case of amplicons). In either ways, libraries are made of these fragments, by adding the appropriate adaptors (the SMRTbell for PacBio, or P5 and P7 for Illumina). The start of the fragment is the part where the sequencer starts to sequence. In most cases, there is no connection to connection to the biology. However, in some cases there is. Most RNAseq protocols these days are stranded, so the start of the read is always according a specific strand. Another example is with amplicons. They will always start with primer 1 and end with primer 2 of the target.
For reads, we have to split the different platforms.
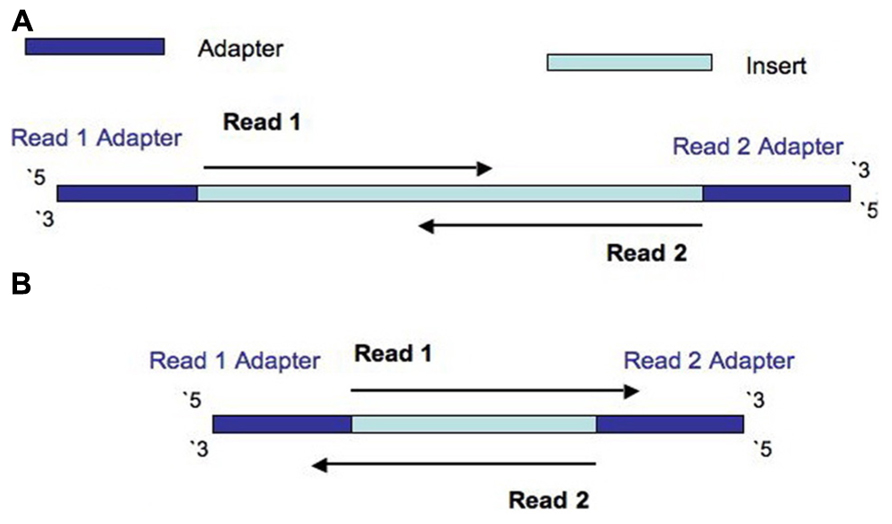
For Illumina, you have single read and paired end sequencing. The single read means that you only sequence the first x bp of the fragment, starting from the start. For paired end, there are 2 reads. The first read, is exactly like the single read sequencing. The second read, starts at the end of the fragment, sequencing x bp towards the start of the fragment (see A on the figure). This has a couple of consequences. Let’s start with the easiest case: RNAseq Differential Expression (DE). In this case you want to compare the genes across conditions. These comparisons are made by counting the number of fragments seen per gene. So, if you sequence paired end, and count the number of reads, you actually count each fragment twice (for DE I always advice only sequencing single reads). For all other cases, I need to introduce a new definition. Fragment size or fragment length, is the length of the fragment (the total number of bases in the fragment, beware that in Bioinformatics, this is the actual insert. In the lab this is usually with the adaptors). If the read length of a read is longer then the fragment, you will read the adaptor (see B on the figure). If the total sequenced bp (the sum of both read lengths) is more then the fragment size, this means that the middle bp will be sequenced twice. This is something you have to consider when analysis NGS data for Illumina platforms. (figure is from this paper)
 For PacBio, you have the subreads and the circular consensus reads (ccs) or reads of insert (ROI). Sequencing happens at the start of the SMRTbell, sequencing the fragment, sequencing the SMRTbell again, sequencing the reverse complement of the fragment, sequencing the SMRTbell again, sequencing the fragment, … (see next figure) The number of times this happens depends on the fragment size, and some other parameters. Each time the sequencer goes over the fragment a subread is created (forward and reverse complement seperated). The PacBio software can create a consensus of the multiple passes over the fragment to create a CCS or ROI. These reads are of high quality. (You can find more about this on the PacBio website).
For PacBio, you have the subreads and the circular consensus reads (ccs) or reads of insert (ROI). Sequencing happens at the start of the SMRTbell, sequencing the fragment, sequencing the SMRTbell again, sequencing the reverse complement of the fragment, sequencing the SMRTbell again, sequencing the fragment, … (see next figure) The number of times this happens depends on the fragment size, and some other parameters. Each time the sequencer goes over the fragment a subread is created (forward and reverse complement seperated). The PacBio software can create a consensus of the multiple passes over the fragment to create a CCS or ROI. These reads are of high quality. (You can find more about this on the PacBio website).
